Modar Tutorial
Example 1: Building a water box
- We'll build a water box from a single water molecule in this exercise.
- And these procedures can actually serve as a general routine for building any solvent box starting from a single unit.
- This example can be found in folder "Example/waterbox" in Modar prebuilt binary package.
- 很抱歉,没时间写中文版了,请大家凑和看,有问题请随时和我联系 chenmengen@gmail.com 。
Here is the procedures step-by-step
Single water molecule in PDB format
- We need to prepare a single water molecule structure file in PDB format, say "tip3.pdb" with following data:
ATOM 1 OH2 WAT 1 0.000 0.000 0.000 ATOM 2 H1 WAT 1 1.080 0.000 0.000 ATOM 3 H2 WAT 1 0.000 1.080 0.000
- For information about PDB foramt, please refer to documentation on RCSB website.
Step 1. Describe the water molecule in Force field
and do a conformation optimization by following command
|
where, "modar" is the primary binary of Modar package,
"step1_single_opt.inp" is the script file in Modar script language,
- Let's take a glance to the script file "step1_single_opt.inp"
where, we use command "loadfftop" and "loadffprm" to load CHARMM 27 force field first; then loaded the the single water by command "loadmsf"; then we call "atom" command to do some nomination modification; then we call "build" command to describe the water molecule in CHARMM forcefiled; then we do a 100 step energy minimization to optimize the water molecular using comand "domin"; finally we use command "savemsf" to store the structure to Modar "msf" format file, and also save the optimized structure a PDB format file.#load force field
loadfftop fmt=chm file="top_all27_prot_na.rtf"
loadffprm fmt=chm file="par_all27_prot_na.prm"
#load molecule structure file
loadmsf fmt="pdb" file="tip3.pdb"
#replace the resdiue name "WAT" to "TIP3" which used in CHARMM force field
atom resname newname="TIP3" select="all"
#assign a segment name
atom segname newname="SOLV" select="all"
#build and describe the molecule according to the force field
build top
#do minimization for the single molecule
domin type=sd nstep=100 nprint=10 ftol=1e-5
#save msf and pdb
savemsf file="step1_single.msf"
savecrd fmt=pdb file="step1_single.pdb"
- The output files will be
Filename
File Type
Description
step1_single_opt.log
Text
The log message produced by Modar, as shown in it that the minimization reached the force tolerance 1e-5 at step 60.
step1_single.msf
Text
Molecular structure file in Modar format with interpretation by the Force Field given
step1_single.pdb
Text
Molecular structure file in RCSB PDB format
Step 2. Generate a water box
by replicating from the single water molecule by command:
|
It will prompt user interactive message for the MSF file, density and the size of box.
Or by follow command without interactive prompt.
|
- Please read the scipt file "step2_genbox.inp" for more details about these options.
- The output files will be:
Filename
File Type
Description
step2_genbox.log
Text
The log message produced by Modar, please read it and the input script if you are fresh to Modar, it may help a lot to understand how Modar works.
step2_solvbox.msf
Text
Molecular structure file in Modar format with interpretation by FF given
step2_solvbox.pdb
Text
Molecular structure file in RCSB PDB format
step2_solvbox_min1.ene
step2_solvbox_min2.ene
step2_solvbox_min3.ene
Binary
The energies profile of energy minimization
pbc.inc
Text
PBC information for further MD simulation
- Right now we have a pseudo-crystal water in a cubic box with total 1131 water molecules.
- Please view the PDB file using VMD or Movar simply by command:
./movar step2_solvbox.pdb - Please view the energy profile by command:
./viewene step2_solvbox_min3.ene
Step 3. Melting at 500k by command:
The box will be heated up to using Berendsen thermostat and equlibrates for 1ns more using Nose-Hoover temperature coupling in NVT ensemble. |
- Please read the scipt file "step3_melt.inp" for more details.
- The output files will be:
Filename
File Type
Description
step3_melting.log
Text
The log file
step3_melting.trj
Binary
Trajectory in Movar binary format
step3_melting.rst
Binary
The MD system state for resuming run
step3_melting.rst.prev
Binary
The “backup” of last “rst” file, Modar make a backup for old restarting state when writing a new state for restarting run to deal some accidents like the storage run out off capacity.
step3_melting.ene
Binary
The energies profile of MD simulation
- The MD timing information would be printed on screen like:
<<Running 2500000 steps simulation with timestep 0.002 ps << Integrator: VVerlet << Ensemble: NVT << Thermostat type: Nose-Hoover << Temperature of reservoir: 500 K << Mass of virtual particle: 1000 << << Time escaped: 0.100591s for 10 steps << MD rate: 8.58924e+06 steps/day, 17.1785 ns/day <======== << Time Left: 6 hours and 59 minutes << << Time escaped: 0.866125s for 100 steps << MD rate: 9.97547e+06 steps/day, 19.9509 ns/day <======== << Time Left: 6 hours and 0 minutes << << Total size of the TRJ file will be about 229.00 Mb for 5.005 ns simulation << << Time escaped: 4.31613s for 500 steps << MD rate: 1.0009e+07 steps/day, 20.0179 ns/day <======== << Time Left: 5 hours and 59 minutes << << Time escaped: 60.3504s for 7500 steps << MD rate: 1.07373e+07 steps/day, 21.4746 ns/day <======== << Time Left: 5 hours and 34 minutes << << Time escaped: 799.044s for 97500 steps << MD rate: 1.05426e+07 steps/day, 21.0852 ns/day <======== << Time Left: 5 hours and 28 minutes
- Please view the trajectory "step3_melting.trj" by command:
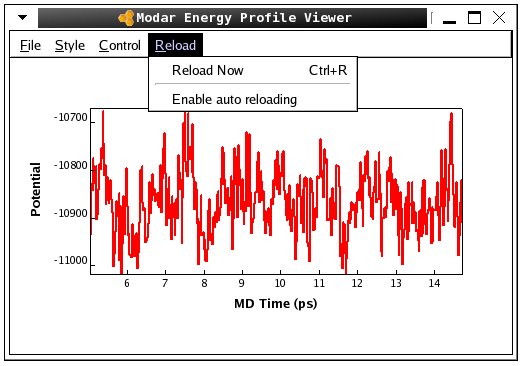
./movar step2_solvbox.pdb step3_melting.trj - Please view the energy profiles "step3_melting" by command:
./viewene step3_melting.eneHere is a snap of the fluctuation of totoal potential energy shown in Modar energies profile "viewene".
Step 4. Equilibration at room temperature by command:
|
- It will run a short annealing to 300K using Bereandsen temperature coupling, and do 2ns equilibation using Nose-Hoover thermostat to relax the system.
- Please read the explanation of the scipt file "step4_equil.inp" for more details.
- Or we may use "nstep=0" and it will build a MD core only stored in file "step4_equil.mds", which is a compact binary file contains all data and parameters for further MD simulation, and then copy the file to a HPC cluster and run the job using a MPI version modar by command:
|
After job done, we need run following script to extract the final structure from the trajectory.
where, we load the "msf" file first, then query the time information of the "trj" file using command "trjin", then load the last step frame of the "trj" file, then call command "img2box" to move all molecules into the primary box, finaly we save the "msf" file for further usage. This script can server as a general template for extracting data from a trajectory file. |
- The output files will be:
Filename
File Type
Description
step4_equil.log
Text
The log file
step4_annealing.trj
Binary
Trajectory fie in Movar format on annealing stage
step4_ annealing.rst
Binary
The MD system state for resuming run
step4_annealing.ene
Binary
The energies profile on annealing stage
step4_equil.trj
Binary
Trajectory file on equilibration stage
step4_equil.ene
Binary
The energies profile on equilibration state
solvbox.msf
Text
The final structure of the water box in Modar MSF format
solvbox.pdb
Text
The final structure in PDB format
- "solvbox.msf" can be used as a raw unit for building a larger water box as shown in other examples.
- Please view the energy profiles by command:
./viewene step4_equil.ene - Please view the trajectory by command:
./movar step2_solvbox.pdb step4_equil.trj
Step 5. Calculate the radial distribution function by command:
|
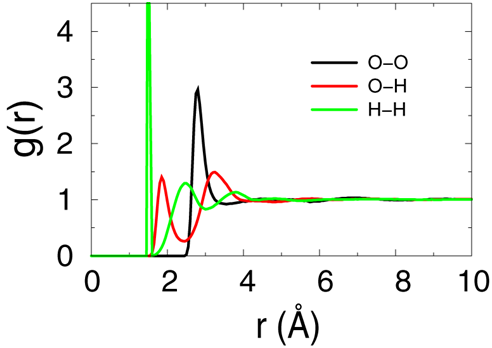
- The raidal distribution function over the last 100ps samples will be evaluated to make sure that we got a well equilibrated water solvent box.
- Please read the explanation of the scipt file "step5_calc_rdf.inp" for more details.
- The output files will be "rdf_O_O.txt", "rdf_O_H.txt" and "rdf_H_H.txt". Please plot this 3files together using xmgr or gnuplut. The result should be simlar to bellow figure, which means the system is equilibrated enough according to reference [Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K, Pekka Mark and Lennart Nilsson,
J. Phys. Chem. A 2001, 105, 9954-9960].
Summary
- In this practice, we generated a small water box with 1331 molecules by replicating from a single water molecule first; Then the pseudo-crystal was heated up to 500K and melted through 1ns NVT simulation; Then the system was annealed to 300K and being equilibrated for 2ns more with Nose-Hoover thermostat; Finally the last 100ps samples were used for radial distribution function (RDF) calculations, and the results indicated the water box is well relaxed.
Computational detail
- The water box was built basing on CHARMM 27 Tip3p model in cubic shape with density of 1.0 g/cm.
- Berendsen weak temperature coupling method were used on both tempering and annealing processes.
- Nose-Hoover thermostat approach was used for keeping constant temperature in all NVT equilibrating simulations.
- All hydrogen relatived bonds were constrained through Shake algorithm in all simulation.
- The long range electrostatic interactions were evaluated precisely through PME algorithm, and the short range interactions were truncated at 10.0 angstrom with a switch function.
- All simulations were propograted using leapfrog integrator with a time step 2fs, and samples were collected for every 1ps second.
Contact us
| Phone: | 400-660-8656 | |
| Email: | support@beemd.org |
我们长期和北京市计算中心合作提供计算培训服务,承接托管计算业务,如有需求请随时联系我们。