Modar Tutorial
Example 3: Ionic liquid simulation
- This example demonstrates how to build ionic liquid solvent from scratch, and then a simple dissolution process will be simulated.
- This example can be found in folder "Example/ils" in Modar prebuilt binary package.
- 很抱歉,没时间写中文版了,请大家凑和看,有问题请随时和我联系 chenmengen@gmail.com 。
Here is the procedures step-by-step
Preparations
- We need to start from a single ions pair 1-n-butyl-3-methylimidazolium hexafluorophosphate [bmim]+[PF6]- (see bellow figure) in PDB format files.
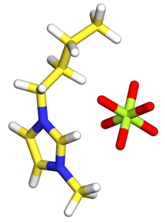
- For information about PDB foramt, please refer to documentation on RCSB website.
- We have the files in the folder at beginning:
Filename
Description
bmi.pdb
The anion structure data file in PDB format
pf6.pdb
The cation structure data file in PDB format
modar
or:
modar.exe (forWindows)The primary Modar executable binary
movar
The structure visulalization tool
viewene
or:
viewene.exe (forWindows)The utility for viewing the energies profile generated by modar
top_all27_prot_na.rtf
The residues’ topologies file of CHARMM force field
par_all27_prot_na.prm
The parameter files of CHARMM force field
bmi.top
pft.top
The ions topology in CHARMM force field format
bmi.prm
pft.prm
CHARMM force field parameters for the ions
Waterbox.msf
The small water box built in Example
Step 1. Build a ions pair by command:
where,
Or, just simply by command if you don't want the interactive prompt.
|
- Let's take a glance to the script file "step1_build_pair.inp"
where, we load the the anion and cation first and then describe the ions in CHARMM forcefiled; and then we use command "atom" to translate the ions, and then a displacement restraint is applied to the ionic pair, finally we do a short energy minimization to refine the position of ionic pair.if(!-var mol1) getvar mol1 msg="no mol1 specified" chkfile=true
if(!-var mol2) getvar mol2 msg="no mol2 specified" chkfile=true
if(!-var dis) getvar dis msg="no displacement distance specified" \
default=6.0 min=1.0 max=20.0
if(!-var nstep) getvar nstep msg="how may cycles for energy minimiztion" \
default=500 min=10 max=1000
#load force field
include "loadff.inc"
#load first molecule
loadmsf fmt="pdb" file=$mol1 build=false
#reset iseq of first molecule to 1
atom iseq newiseq=1 select="all"
tell natom select="all"
natom1=$natom
#load the other molecule
loadmsf fmt="pdb" file=$mol2 append=true build=false
#reset iseq of first molecule to 2
atom iseq newiseq=2 select="iatom >=$natom1"
#change segment name to "ILC"
atom segn newname="ILC" select="all"
#build topology
build top
#replace molecules and separated somehow
atom transto pos=(0,0,0) select="iseq 1"
atom transto pos=($dis,0,0) select="iseq 2"
#add a restrain to pack a closed pair
restrain dist k=100 refvalue=3 select="iseq 1" select="iseq 2"
#do minimization for the single molecule
domin type=sd nstep=$nstep nprint=10 ftol=1e-5
#remove restraint
restrain clear
#save msf and pdb
savemsf file="step1_pair.msf"
savecrd fmt=pdb file="step1_pair.pdb"
- The output files will be
Filename
File Type
Description
step1_build_pair.log
Text
The log message produced by Modar
step1_pair.msf
Text
Ions pair structure file in Modar format with interpretation by FF given
step1_pair.pdb
Text
Ions pair structure file in RCSB PDB format
Step 2. Generate a box by replication from the single pair by command:
|
The script used here is the same as the one used in the step2 of Example 1. The only difference is the feeding in MSF, density, box length, and no argument “rotate=true” needed.
- Please read the scipt file "step2_genbox.inp" for more details about these options.
- The output files will be:
Filename
File Type
Description
step2_genbox.log
Text
The log message produced by Modar, please read it and the input script if you are fresh to Modar, it may help a lot to understand how Modar works.
step2_solvbox.msf
Text
Molecular structure file in Modar format with interpretation by FF given
step2_solvbox.pdb
Text
Molecular structure file in RCSB PDB format
step2_solvbox.ene
Binary
The energies profile of energy minimization
pbc.inc
Text
PBC information for further MD simulation
-
Right now we have a “pseudo-solid” ions box with 250 pairs ions, the box size is 30x30x30; next we’ll melt it at 500K for 1ns MD simulation.
Step 3. Melt the small box by command:
This step is exactly the same as the step 3 in Example 1. But we may need to run longer to reach fully melting state. The MD timing information will be pritned on screen. It took about an hour on my laptop (Intel I5 CPU). |
- Please read the scipt file "step3_melt.inp" for more details.
- The output files will be:
Filename
File Type
Description
step3_melting.log
Text
The log file
step3_melting.trj
Binary
Trajectory in Movar binary format
step3_melting.rst
Binary
The MD system state for resuming run
step3_melting.rst.prev
Binary
The “backup” of last “rst” file, Modar make a backup for old restarting state when writing a new state for restarting run to deal some accidents like the storage run out off capacity.
step3_melting.ene
Binary
The energies profile of MD simulation
Step 4. Equlibrate the small box at room temperature by command:
This step is exactly the same as the step 4 in Example 1. But we may need to run longer since the ions liquid is more viscous than water. |
- Please read the scipt file "step4_equil.inp" for more details.
- The output files will be:
Filename
File Type
Description
step4_equil_nvt.log
Text
The log file
step4_annealing.trj
Binary
Trajectory fie in Movar format on annealing stage
step4_ annealing.rst
Binary
The MD system state for resuming run
step4_annealing.ene
Binary
The energies profile on annealing stage
step4_equil_nvt.trj
Binary
Trajectory file on equilibration stage
step4_equil_nvt.rst
Binary
The MD system state for resuming run
step4_equil_nvt.ene
Binary
The energies profile on equilibration state
- please view the trajectory by command
./movar step2_solvbox.pdb step4_equil_nvt.trj - Here is a snap shot of final state by movar
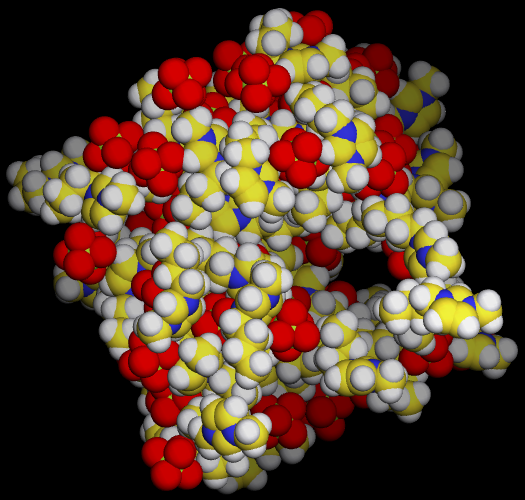
- As shown in the picture, there are some cavities in the system, since we initiated a wrong density in begining. So we need to do a NPT equilibration to get a correct density.
Step 4.1. Equlibrate under NPT ensemble by command:
|
- Please read the scipt file "step4.1_equil_npt.inp" for more details.
- The output files will be:
Filename
File Type
Description
step4_equil_npt.log
Text
The log file
step4_equil_npt.trj
Binary
Trajectory file on equilibration stage
step4_equil_npt.ene
Binary
The energies profile on equilibration state
step4_equil_npt.rst
Binary
The MD system state for resuming run
step4_equil_npt.msf
Text
The final structure in Modar MSF format
step4_equil_npt.pdb
Text
The final structure in PDB format
- Please view the trajectory by command
./movar step2_solvbox.pdb step4_equil_npt.trj - There is no cavity anymore.
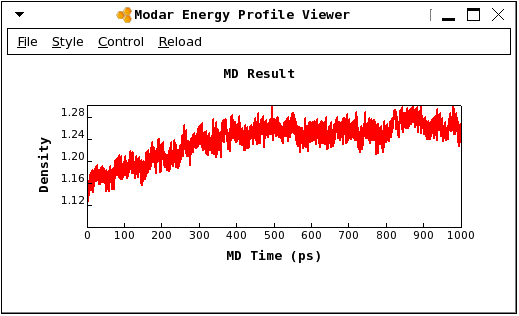
- Please view the energy profile by command
As shown in bellow figure, the system reached the real density at about 500ps../viewene step4_equil_npt.ene
Step 4.2. Calculate the radial distribution function by command:
|
- We do this calculation to make sure the system is equlibrated enough.
- Please read the scipt file "step4.2_calc_rdf.inp" for more details.
- The ions pair RDF will be saved to file “rdf_bmi_pf6.txt”.
- Here is the result should be.
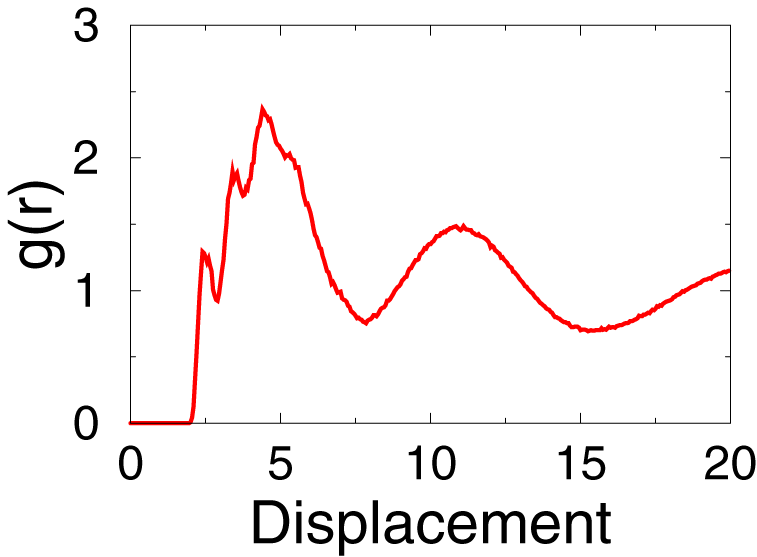
As indicated by the picture, apparently the electrostatic interaction contribution is very significent in ionic liquid simulation.
Step 5. Build a larger box by replicating from the small box by command:
|
Or by following command to generate a cubic box with 500 pairs ions, and the size of box will be determined automatically.
- Please read the scipt file "step5_gen_large_box.inp" for more details.
- The output files will be:
./modar step5_gen_large_box.inp npair=250
Step 6. Equlibrate the larger box at room temperature by command:
|
- In this step, we’ll heatup the system to 300K and run a short NVT equilibration.
- Please read the explanation of the scipt file "step6_equil_nvt.inp" for more details.
- The output files will be:
Filename
File Type
Description
step6_equil_nvt.log
Text
The log message produced by Modar
step6_heatup.trj
Binary
The trajectory file in Modar format for heating up progress
step6_heatup.ene
Binary
The energy profile of the heating up progress
step6_heatup.rst
Binary
The system state for further simulation
step6_equil_nvt.trj
Binary
The trajectory file in Modar format for heating up progress
step6_equil_nvt.ene
Binary
The energy profile of the heating up progress
step6_equil_nvt.rst
Binary
The system state for further simulation
Step 6.1. Run a short NPT equilibration by command:
|
- Please read the explanation of the scipt file "step6.1_equil_npt.inp" for more details.
- The output files will be:
Filename
File Type
Description
step6.1_equil_nvt.log
Text
The log message produced by Modar
step6_equil_npt.trj
Binary
The trajectory file in Modar format for heating up progress
step6_equil_npt.ene
Binary
The energy profile of the heating up progress
step6_equil_npt.rst
Binary
The system state for further simulation
step6_equil_npt.msf
Text
The final structure in Modar MSF format
step6_equil_npt.pdb
Text
The final structure in PDB format
Step 7. Add a water phase by command:
|
-
In this step, we’ll add a water phase to the system.
- Please read the explanation of the scipt file "step7.1_gen_water_box.inp" and "step7.2_merge.inp" for more details.
- The output files will be:
Filename
File Type
Description
step7_add_water.log
Text
The log message produced by Modar
step7_waterbox.mds
Text
step7_waterbox.pdb
Text
step7_two_phases.mds
Text
The whole MD system steup in Modar MDS format file
step7_two_phases.pdb
Text
The whole MD system structure in RCSB PDB format file
step7_two_phases.msf
Text
The final structure in Modar MSF format
-
Please visualize the system by command:
Here is the system look like../movar step7_two_phases.pdb

Step 8. Equilibrate the sytem with boundary restraint by command:
The job will be done in a few minutes, and some timing information will be printed on the screen. |
- Please read the explanation of the scipt file "step8_equil_npt.inp" for more details.
- As shown in the input script file, we added two restraints term to maintain a interface between the two phases.
"restrain zRange" to restrict the ions walking to the water phase;
"restrain zBlock" to block the water moving to the ionic phase;
- The output files will be:
Filename
File Type
Description
step8_equil_npt.log
Text
The log message produced by Modar
step8_equil_npt.mds
Text
The whole MD system structure in Modar MSF format file
step8_equil_npt.trj
Binary
The trajectory file in Modar format for the progress
step8_equil_npt.ene
Binary
The energy profile of the progress
step8_equil_npt.rst
Binary
The system state for further simulation
-
Please view the energy profiles by command:
./viewene step8_equil_npt.eneThere is a extra energy term, "RestraintEe", which is the restraint energy.
- Please view the trajectory by command:
./movar step7_two_phases.pdb step8_equil_npt.trjIt should show a quick meshing event on the phase boundary.
Step 9. Simulate the phases meshing process by command:
Or we could set argument “nstep=0” to build the MD core only, then submit job to a compute node to run job by command:
The job may take a few hours on a laptop, so it’s better to submit the job to a HPC. |
- Please read the explanation of the scipt file "step9_meshing.inp" for more details.
- The output files will be:
Filename
File Type
Description
step9_meshing.log
Text
The log message produced by Modar
step9_meshing.mds
Text
The whole MD system steup in Modar MDS format file
step9_meshing.trj
Binary
The trajectory file in Modar format for the progress
step9_meshing.ene
Binary
The energy profile of the progress
step9_meshing.rst
Binary
The system state for further simulation
-
Please view the energy profiles by command:
./viewene step9_meshing.ene
Step 10. Result analysis by command:
Please wait the step9 to be done properly first. |
- Please read the scipt file "ana.inp" for more details.
- The output files will be:
Filename
File Type
Description
zrgy.txt
Text
The profile of radius of gyration of ionic liquid (only Z component)
aout.txt
Text
How many anion moved to the water with the time going
cout.txt
Text
How many cation moved to the water with the time going
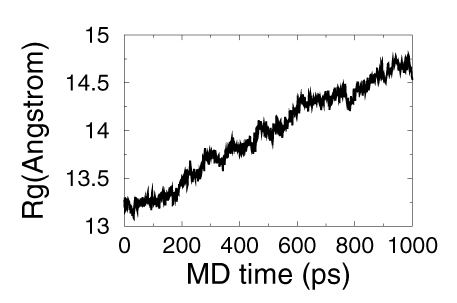
- Here we use the changing of radius of gyration of the ionic liquid as the measurement of the meshing process.
- Here is the how many number of cation and anion moved to the water with the time going
I cannot draw a solid conculation since I don't have enough samples yet. However, it does show us that this ionic liquid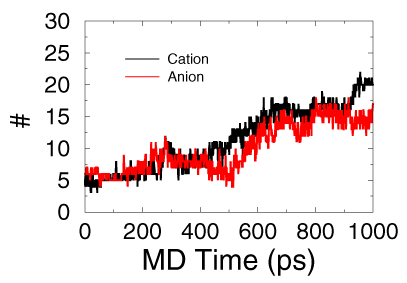
Summary
- In this practice, we built a ionic liquid solvent from scratch, then we add water to the system to create a two phase system, then the meshing process between the ionic liquid and water was simulated.
Computational detail
- The MD system was modeled in CHARMM 27 force field.
- Berendsen weak temperature coupling method were used for tempering process.
- Nose-Hoover thermostat approach was used for keeping constant temperature in NVT and NPT equilibrating simulations, the Langevin piston approach was used for constant pressure coupling.
- Shake constraint method was applied for all hydrogen relatived bonds in all simulation.
- The long range electrostatic interactions were evaluated precisely through PME algorithm, and the short range interactions were truncated at 10.0 angstrom with a switch function.
- All simulations were propograted using leapfrog integrator with a time step 2fs, and samples were collected for every 1ps second.
Contact us
| Phone: | 400-660-8656 | |
| Email: | support@beemd.org |
我们长期和北京市计算中心合作提供计算培训服务,承接托管计算业务,如有需求请随时联系我们。