Modar Tutorial
Example 2: PMF of ions pair: K+ Cl- in water
- This instance can actually serve as a general routine for evaluating the displacement PMF of pair of molecules in solvent.
- This example can be found in folder "Example/kcl_pmf" in Modar prebuilt binary package.
- 很抱歉,没时间写中文版了,请大家凑和看,有问题请随时和我联系 chenmengen@gmail.com 。
Here is the procedures step-by-step
-
Generate a water box by replicating from the small box built in Example 1
-
Continue 5ns more NPT simulation to collect samples for RDF evaluation
Preparations
- We need to start from a single chlorine ion in a PDB format file as shown bellow.
ATOM 1 CLA CLA 1 0.000 0.000 0.000 - For information about PDB foramt, please refer to documentation on RCSB website.
- So let’s make a folder, say “kcl_pmf”, and create a text file “cl.pdb” with above contents, and copy Modar executable binaries, CHARMM force field files and the water box build in Example 1.
- We have the files in the folder at beginning:
Filename
File Type
Description
cl.pdb
Text
The single chlorine anion in PDB format
waterbox.msf
Text
The small water box generated in Example 1
modar
or:
modar.exe (forWindows)Executable binary
The primary Modar executable binary
viewene
or:
viewene.exe (forWindows)Executable binary
The utility for viewing the energies profile generated by modar
top_all27_prot_na.rtf
Text
The residues’ topologies file of CHARMM force field
par_all27_prot_na.prm
Text
The parameter files of CHARMM force field
Step 1. Describe the single chlorine anion in CHARMM FF by following command
|
where, "modar" is the primary binary of Modar package,
"step1_build_solute.inp" is the script file in Modar script language,
- Let's take a glance to the script file "step1_single_opt.inp"
where, we use command "loadfftop" and "loadffprm" to load CHARMM 27 force field first; then loaded the the single chlorine ion by command "loadmsf"; then we call "atom" command to do some nomination modification; then we call "build" command to describe the ion in CHARMM forcefiled; finally we use command "savemsf" to store the structure to Modar "msf" format file, and also save a PDB format file.#load force field
loadfftop fmt=chm file="top_all27_prot_na.rtf"
loadffprm fmt=chm file="par_all27_prot_na.prm"
#load molecule structure file
loadmsf fmt="pdb" file="cl.pdb"
#replace the atom name "CL-" to "CLA" which used in CHARMM force field
atom name newname="CLA" select="all"
#replace the resdiue name "CL-" to "CLA" which used in CHARMM force field
atom resname newname="CLA" select="all"
#assign a segment name
atom segname newname="ION" select="all"
#build and describe the molecule according to the force field
build top
#save msf and pdb
savemsf file="step1_single.msf"
savecrd fmt=pdb file="step1_single.pdb" - The output files will be
Filename
File Type
Description
step1_build_solute.log
Text
The log message produced by Moda
step1_single.msf
Text
Molecular structure file in Modar format with interpretation in force field given
step1_single.pdb
Text
Molecular structure file in RCSB PDB format
Step 2. Generate a box by replication from the small water box created in Example 1
by command:
|
It will prompt user interactive message for the type and the size of box, press enter here to use the default value, here we'll try the truncated octahidralt box which will same some computation cost, the size of 48 Angstrom is good for this instance.
Or by follow command without interactive prompt.
|
- Please read the scipt file "step2_gen_solvent_box.inp" for more details about these options.
- The output files will be:
Filename
File Type
Description
step2_gen_solvent_box.log
Text
The log message produced by Modar, please read it and the input script if you are fresh to Modar, it may help a lot to understand how Modar works.
step2_solvbox.msf
Text
Molecular structure file in Modar format with interpretation by FF given
step2_solvbox.pdb
Text
Molecular structure file in RCSB PDB format
pbc.inc
Text
PBC information for further MD simulation
Step 3. Add more ions by command:
It will prompt the concentration to be. We need add more ions since the PMF is derived from radial distribution function in this example, the default value 0.1M is good for this example. |
- Please read the scipt file "step3_add_ions.inp" for more details.
- The output files will be:
Filename
File Type
Description
step3_add_ions.log
Text
The log message produced by Moda
step3_with_ions.msf
Text
The ions structure in Modar format with interpretation by FF given
step3_with_ions.pdb
Text
The ions structure in RCSB PDB format
Step 3. Put ions into the water box by command:
|
-
We have the water box in step 2 and the ions distribution in step 3; in this step, it shows user how to solvate the ions to the water by merging the two “MSF” together and removing the water molecules which overlaps or have bad contacts with the ions simply.
- Please read the scipt file "step4_merge_all.inp" for more details. It is actually a template for merging two or more parts of MD system together.
- The output files will be:
Filename
File Type
Description
step4_merge_all.log
Text
The log message produced by Modar
step4_all.msf
Text
The whole system in Modar format with interpretation by FF given
step4_all.pdb
Text
The whole sytem in RCSB PDB format file
- please view the MD system build by command
./movar step4_all.pdb - Here is a snap shot by movar
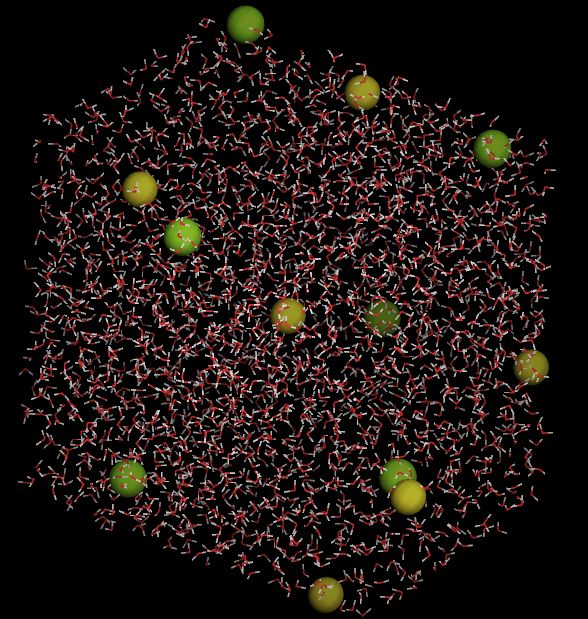
Step 5. Do a energy minimization by command:
The job will be done in a few seconds, and modar will print some timing information on the screen. |
- In this step we’ll do a short energy minimization since the system may still have some bad contacts.
- Please read the explanation of the scipt file "step5_do_minimization.inp" for more details.
- The output files will be:
Filename
File Type
Description
step5_do_minimization.log
Text
The log message produced by Modar
step5_minimized.msf
Text
The whole MD system structure minimized in Modar MSF format file
step5_minimized.pdb
Text
The whole MD system structure minimized in RCSB PDB format file
min?.trj
Binary
The trajectory file in Modar format for every minimization stage respectively
min?.ene
Binary
The energy profile for every minimization state respectively, and it can be viewed by tool “viewene”
miniport.txt
Text
The potential energy profile extracted from the log file for the whole minimization progress.
-
Plot the miniport.txt by xmgr or gnuplot, it should show that the total potential of system is decreased speedly in beginning.
- Or view the energy profiles by command:
./viewene min1.ene
Step 6. Heatup the system by command:
Where “tinit” tells the initial temperature to be generated by Maxwell–Boltzmann distribution, “ttar” is the target temperature, “nstep” tells the total steps for heating. The job will be done in a few minutes, and modar will print some timing information on the screen. |
- In this step, we’ll heatup the system to 300K through Berendsen thermostat approach and equilibrate the system somehow.
- Please read the explanation of the scipt file "step6_heatup.inp" for more details.
- The output files will be:
Filename
File Type
Description
step6_heatup.log
Text
The log message produced by Modar
step6_heatup.mds
Text
The whole MD system steup in Modar MDS format file
step6_heatup.pdb
Text
The whole MD system structure heatuped in RCSB PDB format file
step6_heatup.trj
Binary
The trajectory file in Modar format for heating up progress
step6_heatup.ene
Binary
The energy profile of the heating up progress
step6_heatup.rst
Binary
The system state for further simulation
-
Please view the energy profiles by command:
As shown in bellow pictures, the system has been tempering from 50K to 300K gradually../viewene step6_heatup.ene
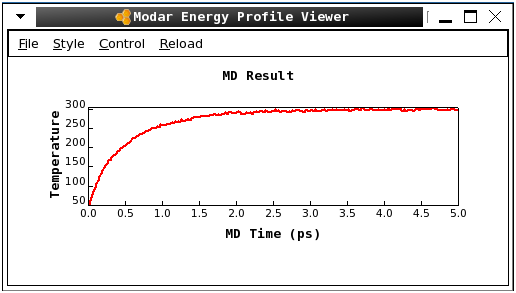
Step 7. Run a short NVT equilibration by command:
The job will be done in a few minutes, and modar will print some timing information on the screen. |
-
In this step, we’ll equilibrate the system under NVT condition with Nose-Hoover thermostat method.
- Please read the explanation of the scipt file "step7_equil_nvt.inp" for more details.
- The output files will be:
Filename
File Type
Description
step7_equil_nvt.log
Text
The log message produced by Modar
step7_equil_nvt.mds
Text
The whole MD system steup in Modar MDS format file
step7_equil_nvt.pdb
Text
The whole MD system structure in RCSB PDB format file
step7_equil_nvt.trj
Binary
The trajectory file in Modar format for equilibrating progress
step7_equil_nvt.ene
Binary
The energy profile of the equilibrating progress
step7_equil_nvt.rst
Binary
The system state for further simulation
-
Please view the energy profiles by command:
Here is a screen shot of the fluctation of temperature../viewene step7_equil_nvt.ene
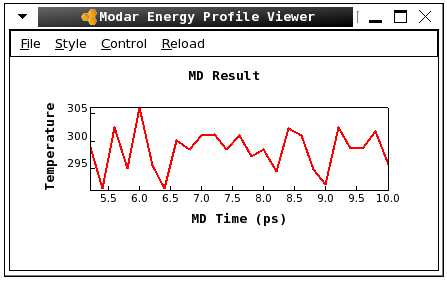
Step 8. Run a short NPT equilibration by command:
The job will be done in a few minutes, and modar will print some timing information on the screen. |
-
In this step, we’ll run a short NPT equilibration with Nose-Hoover thermostat for temperature control and Langevin piston method for pressure coupling. Bellow is the content of script file “step8_equil_npt.inp” to do the equilibration.
- Please read the explanation of the scipt file "step8_equil_npt.inp" for more details.
- The output files will be:
Filename
File Type
Description
step8_equil_npt.log
Text
The log message produced by Modar
step8_equil_npt.mds
Text
The whole MD system structure in Modar MSF format file
step8_equil_npt.pdb
Text
The whole MD system structure in RCSB PDB format file
step8_equil_npt.trj
Binary
The trajectory file in Modar format for the progress
step8_equil_npt.ene
Binary
The energy profile of the progress
step8_equil_npt.rst
Binary
The system state for further simulation
-
Please view the energy profiles by command:
./viewene step8_equil_npt.ene
Step 9. Continue 5ns more NPT simulation by command:
Or we could set argument “nstep=0” to build the MD core only, then submit job to compute node to run job by command:
The job may take a day on a laptop, so it’s better to submit it to a HPC. |
-
We’ll continue the NPT simulation for 5ns more to collect enough samples for PMF evaluation.
- Please read the explanation of the scipt file "step9_equil_npt_more.inp" for more details.
- The output files will be:
Filename
File Type
Description
step9_equil_npt_more.log
Text
The log message produced by Modar
step9_equil_npt_more.mds
Text
The whole MD system steup in Modar MDS format file
step9_equil_npt_more.trj
Binary
The trajectory file in Modar format for the progress
step9_equil_npt_more.ene
Binary
The energy profile of the progress
step9_equil_npt_more.rst
Binary
The system state for further simulation
-
Please view the energy profiles by command:
./viewene step9_equil_npt_more.ene
Step 10. Calculate the radial distribution function by command:
Please wait the step9 done properly first. |
- Please read the explanation of the scipt file "rdf.inp" for more details.
- The output file will be “rdf_K_CL.txt” which is the radial distribution function of K-CL ions pair, the displacement PMF of the ions pairs can be derived simply by following awk command
Plot the file "pmf_K_CL.txt", the PMF should be identical to bellow figure.awk '{ if($2>0) print $1,-0.6*log($2) }' rdf_K_CL.txt > pmf_K_CL.txt
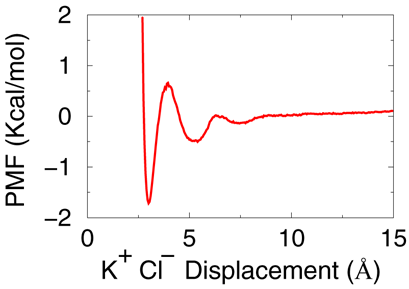
Summary
- In this practice, we generated a truncated octahidralt water box by replicating from the small water box built in Example 1, then we put 0.1M KCL salt to box; then the system was tempered to 300K and undergoed a short NVT equilibration, and also a short NPT equilibration; finally total 5ns second NPT samples were collected for radial distribution function (RDF) evaluation, and displacement PMF of ions pairs was derived.
Computational detail
- The MD system was modeled in CHARMM 27 force field.
- Berendsen weak temperature coupling method were used for tempering process.
- Nose-Hoover thermostat approach was used for keeping constant temperature in NVT and NPT equilibrating simulations, the Langevin piston approach was used for constant pressure coupling.
- Shake constraint method was applied for all hydrogen relatived bonds in all simulation.
- The long range electrostatic interactions were evaluated precisely through PME algorithm, and the short range interactions were truncated at 10.0 angstrom with a switch function.
- All simulations were propograted using leapfrog integrator with a time step 2fs, and samples were collected for every 1ps second.
Contact us
| Phone: | 400-660-8656 | |
| Email: | support@beemd.org |
我们长期和北京市计算中心合作提供计算培训服务,承接托管计算业务,如有需求请随时联系我们。